Radmap
A program for visualizing non linear potential energy surfaces
RADMAP
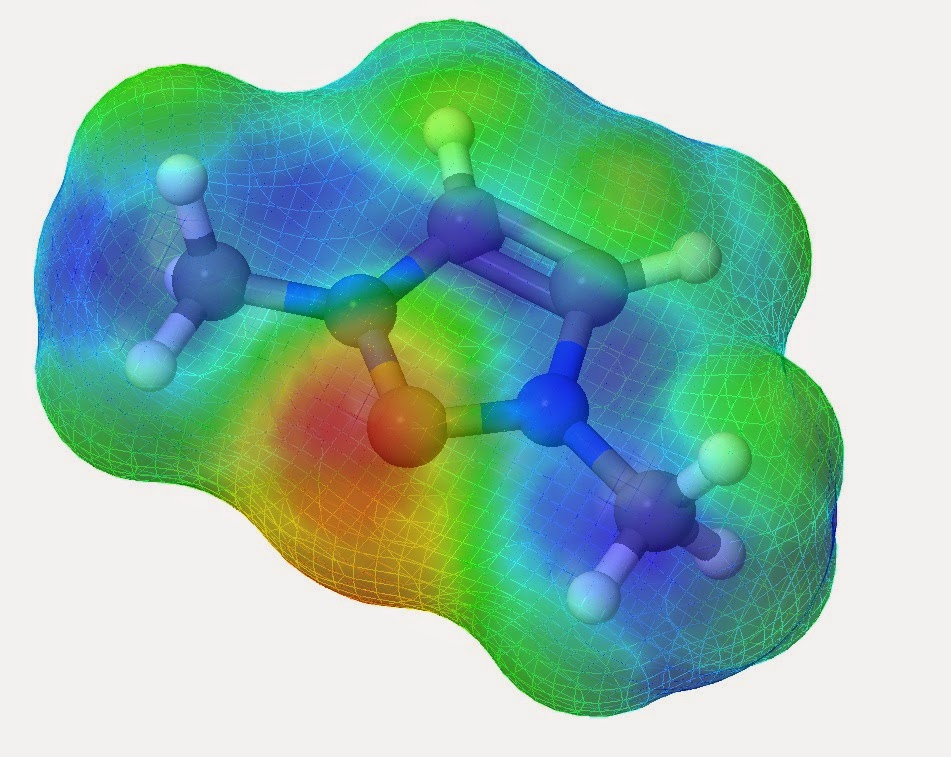
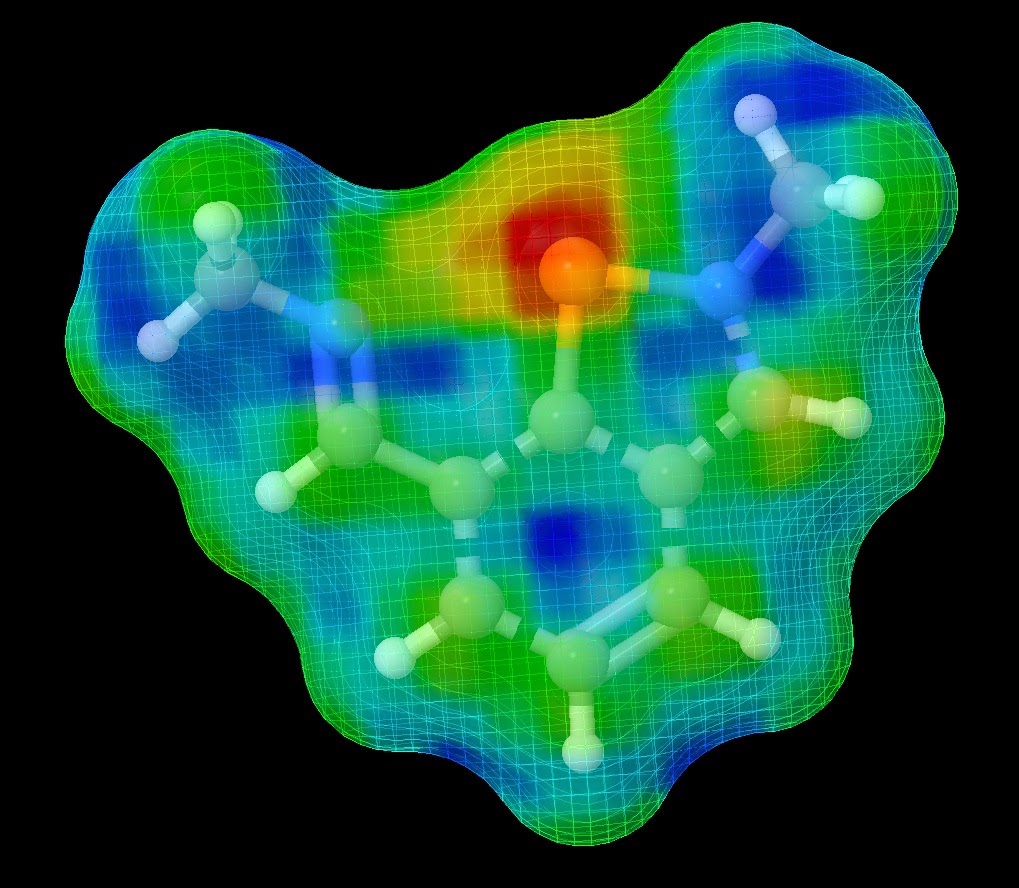
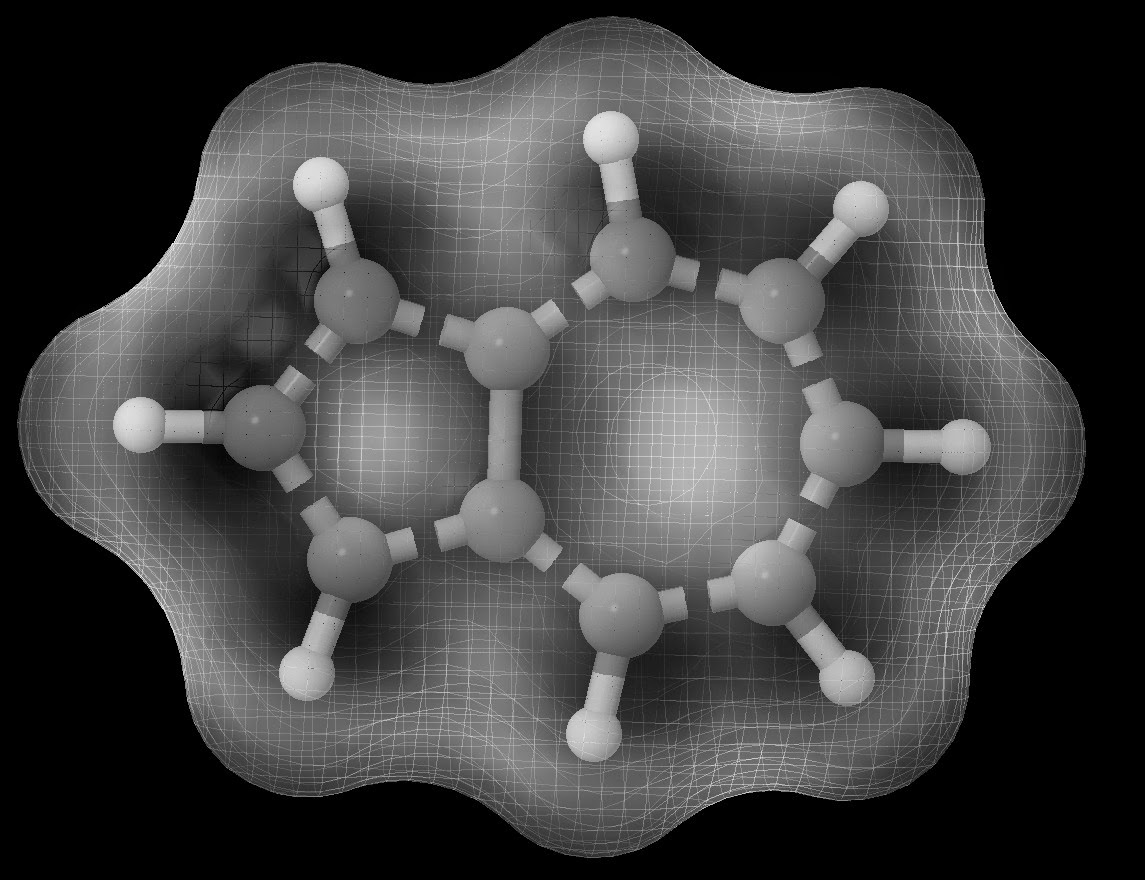
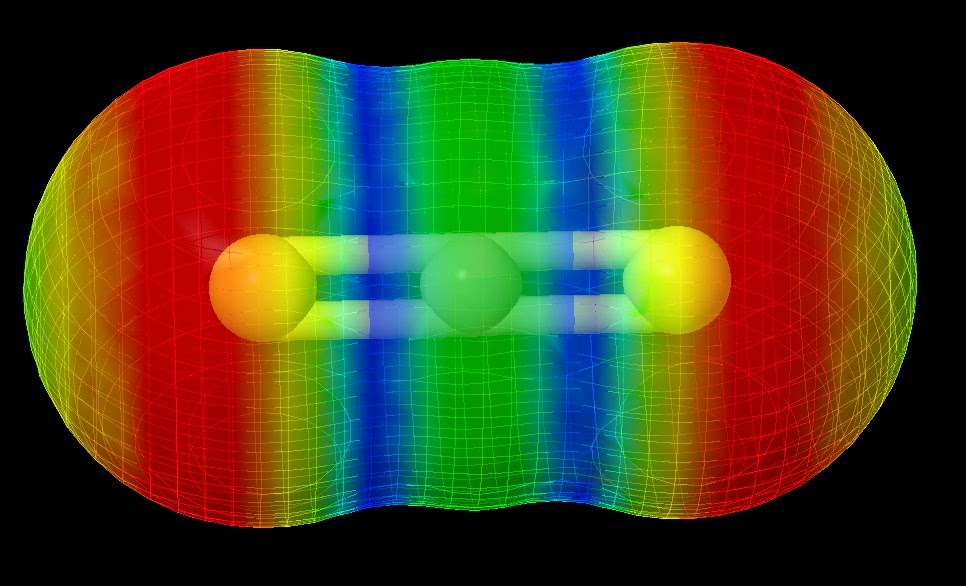
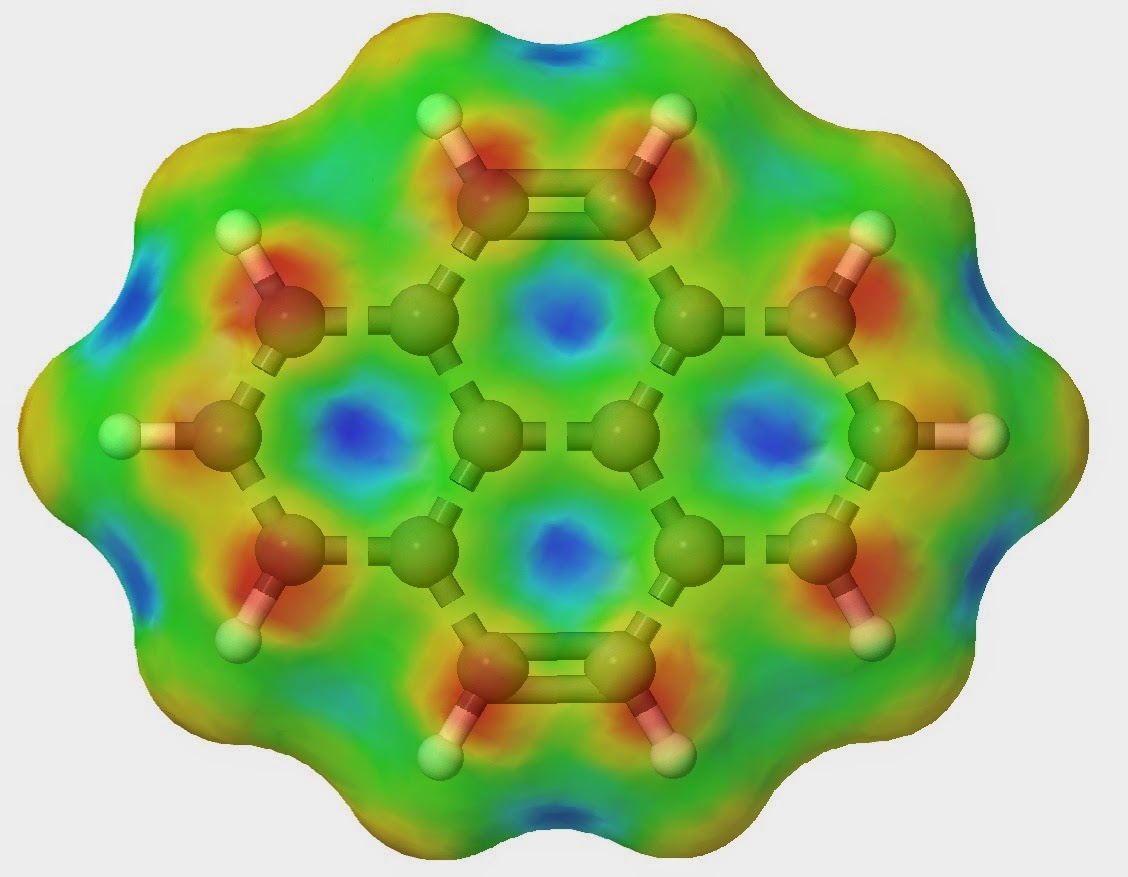
Determining the reactivity of novel compounds is oftentimes a laborious and costly process, which would typically involve a large amount of time in the lab. RADMAP provides a convenient way of visualizing the reaction surfaces of molecules via a purely ab-initio method with out requiring any lab time. To accomplish this the RADMAP program produces an isosurface potential energy surface on the molecule of interest and calculating the energy of all possible aducts formed on this reaction surface. This provides for a quick and convenient look into the earliest possible stages of a reaction.
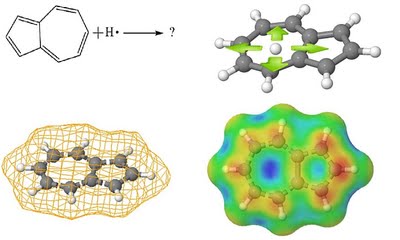
These surfaces provide an intuitive way to view the reactivity of molecules without the need of any lab work. Furthermore, This process has been found to be particularly useful for novel ligands to be attached to metal centers.
The simple interface allows the user to quickly import a .mol file containing the structure of the molecule of interest. Here, a simple control panel allows the user to select which computational method to use and how detailed the mesh should be, once this is finished the program generates a set of input files and a batch file that can be run by the Gaussian suite of computational chemistry programs. Once all the adducts have been calculated, the program collects all of the output files and generates the surfaces seen above. More information will be available with the release of the publication.
 ascorbic acid base v3 white
Main Developer: Andrew K Long
Adviser: Jason A.C. Clyburne and Cory Pye
Art Design: Emily Rose
ascorbic acid base v3 white
Main Developer: Andrew K Long
Adviser: Jason A.C. Clyburne and Cory Pye
Art Design: Emily Rose
Acknowledgement: The authors would like to thank Robert M. Hanson and all the contributors to the Jmol-Development mailing list for their help with this program. Additionally we would like to thank Jason Masuda for his input during the development process.